Classical Molecular Dynamics Series [Part - 4a]: Let us setup a simulation and run it!
Hi guys! After a long gap, @dexterdev is back with the fourth part of Classical Molecular Dynamics (CMD) series. Today let us set up a simulation system in a software called NAMD. For those who missed my initial posts related to CMD, please see it in links below:
Part 1 (The Fundamentals)
Part 2 (The Force Field!)
Part 3 (Solving the molecular dynamics equation)
One thing to be careful about this article is that this is a HOW TO tutorial, rather than explaining concepts. So if you guys are confused about certain technical terms etc, don't worry. I will be explaining them in the future parts. Also, you can ask me doubts in comments, and I will try answering those. :)
What is NAMD?
NAMD is a Molecular Dynamics software developed from Theoretical and Computational Biophysics group from University of Illinois at Urbana-Champaign.
Quoting from their website:
NAMD, recipient of a 2002 Gordon Bell Award, is a parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems.
This group has also designed a software called VMD to visualize the simulations etc.
Firstly, you may want to download NAMD and VMD from this webpage: http://www.ks.uiuc.edu/Development/. Mine is a 64-bit Linux machine. Yours may be a different one. So be careful in choosing your selection. You may also need to register with the website to download these files. But they are all free software.
[ONE THING TO BE CAREFUL: Depending on the size of the molecule and length of the simulation, most of the simulation cases are only possible on high-performance computers like GPU machines/parallel processor machines with multiple cores. Like I use 128 or 256 cores for a job in our Institute cluster facility. So in practice, you can only check if your simulation is running or not by using your desktop machine/laptop.]
Let us do a simulation first!
I am a fan of learning by doing. So let us begin straight away. The example system which we will set up today is that of a phospholipid membrane (specifically DMPC one) in water. To know what DMPC membrane is, please see one of my previous articles:
Phospholipids: The unsung heroes in biomolecules!
So to start the simulation of molecules, we need initial structures, right? We are going to initiate animation in the dead structures based on the laws of physics(basically Newton's second law). The force-field which we are going to use are CHARMM36 version.
Intial structure
For this example tutorial, we are considering a phospholipid membrane (DMPC) in water. So the initial structures are in the forms of PDB and PSF formats. I will explain what those are. PDB structure basically contains the coordinate information of atoms in the molecule.
So we need structure. In the case of bilayer lipid membranes, we can go to CHARMM-gui membrane builder website. Let us select our options there:
- Check the Membrane only system option, first.
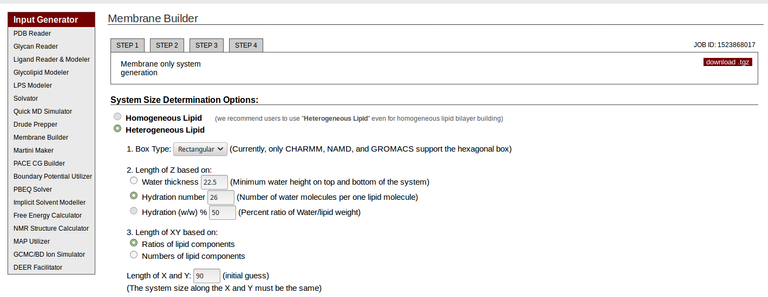
- And click on it. You will be directed here. Please see the below options for screenshots. You should choose Heterogeneous Lipid option first, then Hydration number as 26, Length of XY based on:
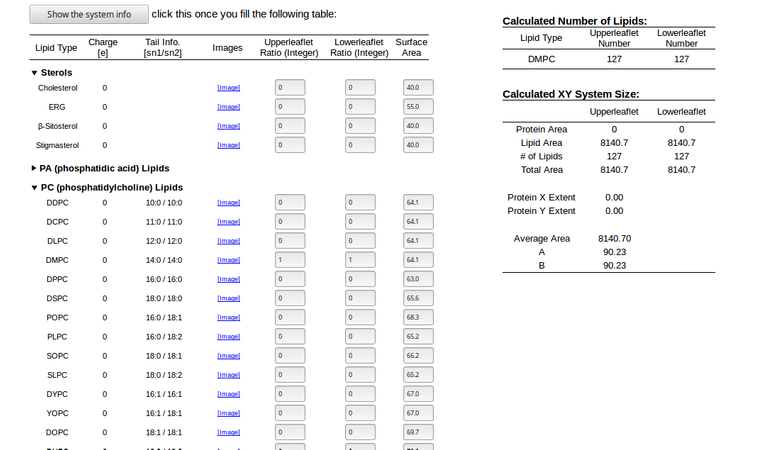
Ratios of lipid components, Length of X and Y:90(angstrom units=10-10 metres, you can choose your length instead of 90) and finally select the DMPC lipid from PC option with 1:1 ratio(which will incorporate 127 upper and 127 lower lipids).
A screenshot from CHARMM-gui website showing the options to choose
Click the next step. I am changing the IONS option to 0.15 Molar/liter NaCl from KCl. But it doesn't matter much. You can see the xyz dimensions are approximately 90 x 90 x 58 Angstrom3. Click next again. (CHARMM-gui will add water and ions on top and bottom of lipid bilayer. )
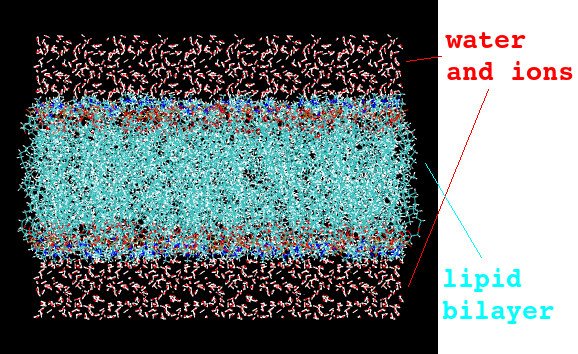
And download the output files in .tgz format. You can see a bunch of files in it. There is also PDB file in it. Let us open it using a text editor and VMD software. Visualize the step4_lipid.pdb (which you got from .tgz file after the last step) output in VMD. You can see something similar to below(DMPC lipid bilayer):

vmd step4_lipid.pdb in terminal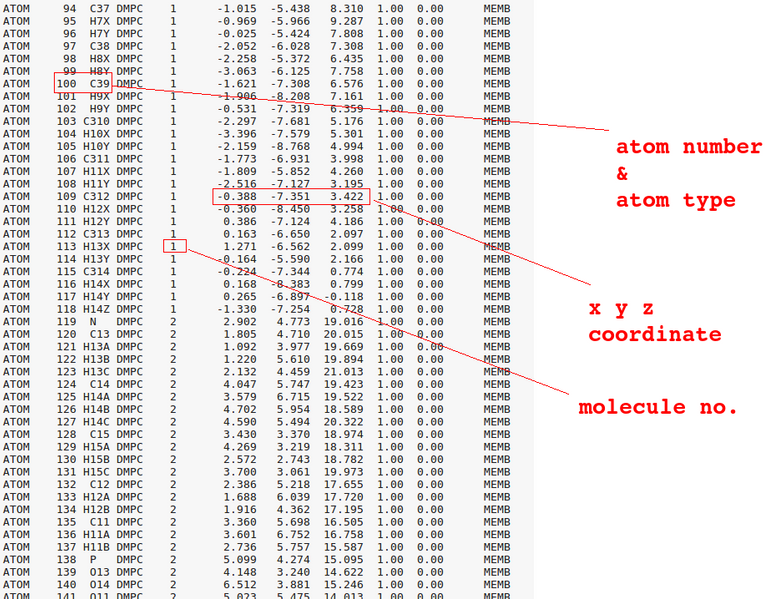
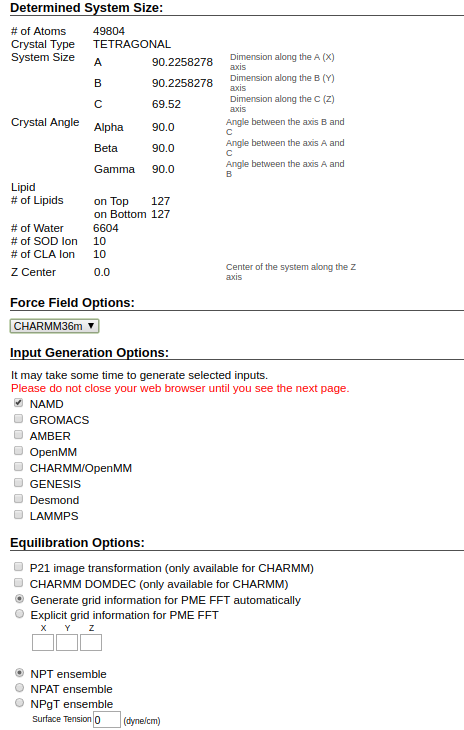
- Choose the options as in below snapshot. We will go through the concept of ensemble etc later. For now, we are going to choose NPT ensemble (Constant number of particles, pressure, and temperature). Forcefield is selected as CHARMM36m(m for modified). Temperature T=303.15.
A note about other file types: PDB file has the initial coordinate information. PSF file will have the atom's charge, mass, and structure related information etc. Besides PSF and PDB files, we have following files:
- topology file(.rtf file) in order to create PSF file from PDB file. (this step is not needed for this particular example since CHARMM-gui creates PSF file for you.)
- parameter file(.prm file) in order to specify parameter values related to bonds, angles, dihedrals, improper dihedrals and Lenard-Jones non-bonding interactions. (You may see my previous MD articles to know what I meant by dihedrals, Lenard-Jones potential etc)
- configuration file where we will input all necessary arguments for the simulation including above-mentioned files too.
So I want to tell you guys one more thing. If the above file types and other details seem daunting for you, let me assure you that we will come to each of these things again in future. Let me remind you that the focus of this article is just to get start doing simulations.
A typical CONFIGURATION file
##############################################################
## JOB DESCRIPTION ##
#############################################################
# DMPC patch equilibration (intitial structure from CHARMM-gui)
#############################################################
## ADJUSTABLE PARAMETERS ##
#############################################################
# structure and coordinate files are PDB and PSF file generated by CHARMM-gui
structure ./step5_assembly.psf
coordinates ./step5_assembly.pdb
set temperature 303.15
set outputname DMPC_patch_NPT
#DCD and other files output name
firsttimestep 0
#############################################################
## SIMULATION PARAMETERS ##
#############################################################
# Input (CHARMM force-field ON)
paraTypeCharmm on
parameters ./par_water_ions.prm
parameters ./par_all36_lipid.prm
temperature $temperature
# Force-Field Parameters (will discuss in future)
exclude scaled1-4
1-4scaling 1.0
cutoff 12.
switching on
switchdist 10.
pairlistdist 15.0
margin 8.0
# Integrator Parameters
# fullElectFrequency*timestep <=4.0
# stable time steps:
# with rigidBonds=all: electro:6fs;short-rangeNB:2fs;bonded:2fs
# otherwise : 2fs 2fs 1fs
timestep 2.0 ;# 2fs/step (our time step is 2 femto second)
rigidBonds all ;# Needed for 2fs steps
useSettle on
nonbondedFreq 1
fullElectFrequency 2
stepspercycle 10
#########################
zeroMomentum yes
#########################
# Constant Temperature Control (Thermostat settings to keep fixed temperature)
langevin off ;# do langevin dynamics
langevinDamping 5 ;# damping coefficient (gamma) of 5/ps
langevinTemp $temperature
langevinHydrogen off ;# don't couple langevin bath to hydrogens
#temperature coupling for temperature coupling
tCouple on
tCoupleTemp $temperature
if {1} {
# Periodic Boundary Conditions (specify the dimensions)
# center of the periodic box; NOT ORIGIN!!!
cellBasisVector1 101.5 0 0
cellBasisVector2 0 101.5 0
cellBasisVector3 0 0 71
cellOrigin 0 0 0
}
# The above numbers are approximated from the data we analysed through VMD.
wrapAll on
# PME (Particle mesh ewald for effiecient calculation of full-system electrostatic calculations in periodic boundary conditions)
# multiples of 2,3,5 & >=dimensions above
PME yes
PMEGridSizeX 120
PMEGridSizeY 120
PMEGridSizeZ 90
# Constant Pressure Control (variable volume)
useGroupPressure yes ;# needed for rigidBonds
useFlexibleCell no
useConstantArea no
langevinPiston on
langevinPistonTarget 1.01325 ;# in bar -> 1 atm
langevinPistonPeriod 100.
langevinPistonDecay 50.
langevinPistonTemp $temperature
# Output
outputName $outputname
restartfreq 500000 ;# every 1 ns(capturing restart points if at all the simualtion breaks, so that you can restart from this point)
dcdfreq 10000 #(capture MD trajectory data with frequency 10000 steps)
xstFreq 10000
outputEnergies 10000
outputPressure 10000
#############################################################
## EXECUTION SCRIPT ##
#############################################################
# Minimization (to take the structure to a local minima using conjugate gradient method)
minimize 5000
reinitvels $temperature ;#Reinitialize velocities(temperature is dependent on velocities of atoms)
run 10000000 ;# run the simulation for 20 ns
Now the task of NAMD software is to run this config file. For this purpose create a folder with necessary files in it. Like PDB, PSF, PRM and restart-files(necessary in case the simulation is restarted from a previous run input).
I know that there are a lot of missing points here. There are unclear things in the config file. But my soul aim in this article is to get started with NAMD.
The files par_all36_lipid.prm and par_water_ions.prm are available from here in compressed formats. But for convenience you can get it from here and here.
Running the config file
devt@BUSYBOX ~/home/devt/SIM $ namd2 DMPC_patch_NPT_eq.config > log
Below is a line from the log file generated:
Info: Initial time: 1 CPUs 0.437884 s/step 2.53405 days/ns 211.227 MB memory
From the log file, we can read that it takes 2.5 days for 1ns with 1CPU approximately! This is the case with 49805 atoms in the system.(We can get this number of total atoms either by querying using VMD software or opening the PDB file in a text editor and inspecting it.) We can improve this by using HPC(high-performance computer) machines.
What this article will enable you to do?
This article simply allows you to see how a simulation run can be realized using NAMD. In the coming parts, we will explore some simulations of real systems like proteins etc by reproducing results from some research papers etc.
Coming up in the next part...
- A detailed description of each filetype including generated output files
- More VMD and NAMD techniques/details
- How can we run the simulations faster?(if there is an access to HPC)
- Visualizing the simulation results and doing some basic analysis etc
- Periodic Boundary Conditions, Neutralizing total charge in the system etc
- A small system, which anyone can try out in their machine
- And more!
References [Books for further reading]
Textbook references for learning theory of Molecular Dynamics:
- "Statistical Mechanics: Theory and Molecular Simulations" by Mark E. Tuckerman
- "Molecular Modelling: Principles and Applications" by Andrew R. Leach
- "Computer Simulation of Liquids" by D. J. Tildesley and M.P. Allen
References specific to NAMD and VMD:
CHARMM-gui related papers:
Join #steemSTEM

And to steemSTEM beginners:

Follow me @dexterdev
____ _______ ______ _________ ____ ______
/ _ / __\ \//__ __/ __/ __/ _ / __/ \ |\
| | \| \ \ / / \ | \ | \/| | \| \ | | //
| |_/| /_ / \ | | | /_| | |_/| /_| \//
\____\____/__/\\ \_/ \____\_/\_\____\____\__/
Wow that is pretty awesome @dexterdev. I thought Blender was pretty tough on CPU, how long did the video above take to generate?
@terrylovejoy : The video I generated above is using a desktop snapshot software. There are better ways to do it using VMD. But this was a dirty hack. :( But I will capture a nice video once. My institute machine is about to explode. It is full of molecular dynamics data and tonnes of installed software. Once I clean my machine, I will start doing it.
Congratulations @dexterdev! You have completed some achievement on Steemit and have been rewarded with new badge(s) :
Click on any badge to view your own Board of Honor on SteemitBoard.
To support your work, I also upvoted your post!
For more information about SteemitBoard, click here
If you no longer want to receive notifications, reply to this comment with the word
STOPHello @dexterdev , I was designed to give advice to "steemit" users.
I recommend to increase this;
The most winning bid bot in the last 24 hours is ✅ "smartsteem"
You can enter "steembottracker.com" to find more offers.
You can make "Resteem" and advertise to the followers of the whale accounts.
"Resteem Bot" for you;
✅ @byresteem has 25.500 Followers + 7000 Sp + Upvote with min +55 accounts.
I am a bot, I can not answer the comment. I hope I could help. Good luck.
That's some horsepower. Instead of simulating, mine btc with it and buy a bigger simulator 😄
You can do both with GRIDCOIN I guess :)
Oh duh 😅